The relationship between cholesterol and heart attack risk is one of the most extensively studied associations in medicine — and one of the most misunderstood by the public. Most people know, broadly, that high cholesterol is bad for the heart. Far fewer understand exactly how it causes a heart attack, what specific numbers carry specific risks, or why some people with apparently “normal” cholesterol still have heart attacks.
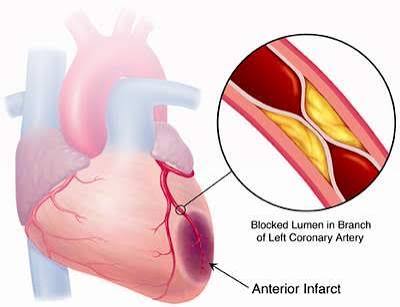
A heart attack — myocardial infarction — is almost always the culmination of a process that began decades earlier. Elevated LDL cholesterol drives the slow, silent accumulation of atherosclerotic plaque within coronary artery walls. The plaque grows, the artery wall becomes inflamed, and eventually an unstable plaque ruptures — exposing its contents to flowing blood, triggering a clot that suddenly and completely blocks coronary flow. Heart muscle downstream, deprived of oxygen, begins to die within minutes.
Understanding cholesterol and heart attack risk means understanding this process: where LDL fits in the causal chain, what your specific cholesterol numbers translate to in terms of probability, and what the evidence says about how to reduce that probability over a lifetime.
How Cholesterol Causes Heart Attacks
The pathway from elevated LDL to myocardial infarction is well characterized. It begins in the arterial wall.
LDL particles — lipoproteins carrying cholesterol through the bloodstream — cross the endothelium (the inner lining of arteries) and enter the arterial wall. When LDL is elevated or the endothelium is injured (by hypertension, smoking, high blood glucose), more particles enter the wall and accumulate. There, they are modified — oxidized — and become highly inflammatory. The immune system responds: macrophages engulf the oxidized LDL and, overwhelmed, become lipid-laden foam cells. Foam cells accumulate into fatty streaks, then into more complex fibrous plaques with a necrotic lipid core covered by a fibrous cap.
Most people understand atherosclerosis as a process of gradual narrowing — pipes getting clogged. That is only part of the picture. The more dangerous event is not gradual narrowing but plaque rupture.
An atherosclerotic plaque with a thin fibrous cap — weakened by macrophage-secreted enzymes — can rupture suddenly, exposing the thrombogenic contents of the lipid core to the bloodstream. Platelets rush to the site, the coagulation cascade activates, and a blood clot (thrombus) forms rapidly. If the thrombus occludes the artery completely, blood flow to the downstream heart muscle stops. Cardiac cells begin dying within 20 minutes of complete ischemia. This is a heart attack.
One of the most important clinical insights in modern cardiology is that the plaques most likely to rupture are not necessarily the most obstructive. Many fatal heart attacks occur at plaques that were narrowing the artery by only 40 to 70 percent — not the large, calcified plaques that show up most dramatically on angiograms. The vulnerable plaque is smaller, softer, more inflamed, and covered by a thinner cap. This is why preventing MI requires long-term control of LDL to slow plaque formation and stabilize existing plaques — not just opening arteries that are visibly narrowed. For a detailed breakdown of how plaque forms, see cholesterol and plaque buildup in arteries.
Cholesterol and Heart Attack Risk — What Specific Numbers Mean
LDL cholesterol (LDL-C) is the primary measure of atherogenic lipid burden and the standard clinical target for heart attack risk reduction. The standard categorization is:
- Below 100 mg/dL: optimal
- 100–129 mg/dL: near/above optimal
- 130–159 mg/dL: borderline high
- 160–189 mg/dL: high
- 190 mg/dL and above: very high (often indicates familial hypercholesterolemia)
Each step up this ladder corresponds to meaningfully higher cardiovascular risk. Data from the Cholesterol Treatment Trialists’ Collaboration — a meta-analysis of major statin trials involving more than 174,000 patients — established that each 1 mmol/L (approximately 39 mg/dL) reduction in LDL-C is associated with roughly a 22 percent relative reduction in major cardiovascular events (heart attack, stroke, coronary revascularization, cardiovascular death). This relationship is consistent across age groups, sexes, baseline LDL levels, and presence or absence of prior cardiovascular disease.
Crucially, the relationship between LDL and cardiovascular risk is log-linear — there is no clearly safe floor. Observational data and Mendelian randomization studies consistently show that lower LDL is associated with lower risk, even at levels below 70 mg/dL. Genetic variants causing lifelong lower LDL are associated with markedly lower lifetime cardiovascular event rates — roughly 2 percent reduction per 1 mg/dL of lifelong lower LDL exposure. For more on what LDL means and how it differs from HDL, see LDL vs HDL cholesterol.
HDL cholesterol (HDL-C). Low HDL — below 40 mg/dL in men, below 50 mg/dL in women — is associated with higher cardiovascular risk and is considered a risk marker by current guidelines. However, pharmacologically raising HDL (niacin, CETP inhibitors) consistently failed to reduce cardiovascular events in clinical trials, suggesting low HDL is a marker of broader metabolic risk rather than a primary therapeutic target in isolation.
Triglycerides. Elevated triglycerides — above 150 mg/dL — are associated with increased cardiovascular risk. Triglyceride-rich lipoproteins are atherogenic; high triglycerides promote small dense LDL formation and are a marker of insulin resistance and metabolic syndrome. The REDUCE-IT trial demonstrated that 4 grams per day of icosapentaenoic acid (EPA) reduced major cardiovascular events by 25 percent in statin-treated patients with elevated triglycerides — establishing elevated triglycerides as a modifiable cardiovascular risk factor beyond LDL.
ApoB (apolipoprotein B) measures the number of atherogenic lipid particles rather than the amount of cholesterol they carry. Since each LDL, VLDL, IDL, and Lp(a) particle contains exactly one ApoB molecule, ApoB directly counts the particles that can penetrate the endothelium. Research shows that ApoB predicts cardiovascular events more accurately than LDL-C when the two measures diverge — particularly in people with metabolic syndrome. Optimal ApoB is below 90 mg/dL; below 65 mg/dL in high-risk patients.
Lipoprotein(a) — Lp(a) is a genetically determined lipid particle not captured in the standard lipid panel. Elevated Lp(a) (above 50 mg/dL, or 125 nmol/L) independently increases heart attack risk and explains many cases of premature coronary artery disease in people who appear to have otherwise normal cholesterol. Lp(a) is almost entirely determined by genetics and does not respond meaningfully to diet or exercise. A one-time Lp(a) measurement is increasingly recommended for all adults, particularly those with premature cardiovascular disease or a family history of early MI.
Can You Have a Heart Attack With Normal Cholesterol?
This question comes up often — and the answer is yes, for reasons that are important to understand.
“Normal” cholesterol on a standard lipid panel does not mean zero plaque, zero inflammation, or zero heart attack risk. It means the rate of LDL-driven plaque formation is lower than at higher LDL levels. But other factors can still produce significant cardiovascular risk:
Other lipid abnormalities not captured by LDL-C. A person with LDL of 110 mg/dL but highly elevated Lp(a) may have substantially higher heart attack risk than someone with LDL of 140 mg/dL and undetectable Lp(a). Similarly, low HDL combined with high triglycerides (the atherogenic dyslipidemia of metabolic syndrome) can drive cardiovascular risk even when LDL appears acceptable.
Other risk factors that amplify risk at any LDL level. Cigarette smoking, uncontrolled hypertension, and diabetes each dramatically accelerate atherosclerosis and plaque vulnerability. A smoker with LDL of 110 mg/dL may have higher cardiovascular risk than a non-smoker with LDL of 145 mg/dL, depending on the magnitude and duration of other risk factors.
Accumulated subclinical atherosclerosis. A single cholesterol test at age 55 does not capture 35 years of exposure between ages 20 and 55. Someone whose LDL was 155 mg/dL throughout their twenties, thirties, and forties may have substantial existing plaque not reflected in a cholesterol reading at 55 that has since normalized through dietary change.
Acute plaque triggers. Emotional stress, cocaine use, and extreme cold exposure can trigger coronary artery spasm or plaque erosion in vessels with pre-existing atherosclerosis — causing MI even at modest LDL levels.
The INTERHEART study — a large international case-control study of first heart attacks in 52 countries — found that approximately nine modifiable risk factors accounted for over 90 percent of the population-attributable risk of MI. Because multiple factors interact and accumulate over time, many individuals who have a heart attack did not appear clearly high-risk on any single parameter before the event. “Normal cholesterol” warrants continued monitoring and attention to the full risk factor picture — not dismissal.
How Doctors Calculate Your Heart Attack Risk
Clinical cardiovascular risk assessment has moved beyond simple cholesterol thresholds to probabilistic models that integrate multiple risk factors simultaneously.
The ACC/AHA Pooled Cohort Equations are the primary guideline-endorsed tool for estimating 10-year risk of a first atherosclerotic cardiovascular event. The calculator uses: age, sex, race/ethnicity, total cholesterol, HDL-C, systolic blood pressure, whether blood pressure is being treated, presence of diabetes, and smoking status. The output is a 10-year probability of first heart attack or stroke:
- Below 5%: low risk — lifestyle modification; statin not generally recommended unless LDL ≥ 190 mg/dL
- 5–7.5%: borderline risk — shared decision-making; lifestyle first; statin appropriate if risk enhancers present
- 7.5–20%: intermediate risk — moderate-intensity statin recommended; consider CAC score if decision is uncertain
- 20% and above: high risk — high-intensity statin recommended; LDL target below 70 mg/dL
The Pooled Cohort Equations have important limitations: they do not incorporate ApoB, Lp(a), coronary artery calcium, family history of premature ASCVD, or inflammatory markers — all of which can meaningfully change individual risk. Risk enhancers that can push borderline-risk patients toward treatment include: family history of premature ASCVD (first-degree relative with MI or stroke before age 55 in men / 65 in women); persistently elevated LDL-C ≥ 160 mg/dL or non-HDL-C ≥ 190 mg/dL; elevated Lp(a) ≥ 50 mg/dL; elevated ApoB ≥ 130 mg/dL; hs-CRP ≥ 2 mg/L; ABI below 0.9; chronic kidney disease; and metabolic syndrome.
The Coronary Artery Calcium (CAC) score has emerged as the most powerful tool for reclassifying risk where calculated risk is uncertain. CAC detects calcified plaque directly with a low-dose CT scan. A CAC score of 0 is associated with very low 10-year cardiovascular event rates even in intermediate-risk patients, supporting deferral of statin initiation. A CAC score of 300 or higher (or above the 75th percentile for age, sex, and race) supports high-intensity statin therapy even if calculated 10-year risk appears moderate.
LDL Targets and When Medication Is Recommended
Treatment decisions for elevated cholesterol center on LDL targets that vary by cardiovascular risk tier:
LDL ≥ 190 mg/dL — high-intensity statin therapy is recommended regardless of calculated 10-year risk. This threshold triggers treatment because LDL this high typically reflects genetic hypercholesterolemia and carries substantial cumulative lifetime risk that a 10-year calculator underestimates.
Established cardiovascular disease (secondary prevention) — the goal is LDL below 70 mg/dL. High-intensity statin therapy is standard. If LDL remains above 70 mg/dL, ezetimibe should be added; if still above target, a PCSK9 inhibitor is indicated.
Very high-risk cardiovascular disease (multiple ASCVD events, or one event plus diabetes/CKD/familial hypercholesterolemia) — LDL below 55 mg/dL is recommended. Combination therapy (statin + ezetimibe ± PCSK9 inhibitor) is often required to reach this target.
Statins inhibit HMG-CoA reductase, reducing hepatic cholesterol production and upregulating LDL receptors; they reduce LDL by 30 to 60 percent depending on statin and dose and also stabilize existing plaques through anti-inflammatory effects. Ezetimibe inhibits intestinal cholesterol absorption; added to a statin, it reduces LDL by an additional 15 to 25 percent and was proven to reduce cardiovascular events in IMPROVE-IT. PCSK9 inhibitors (evolocumab/Repatha, alirocumab/Praluent) are injectable monoclonal antibodies that reduce LDL by 50 to 60 percent additional on top of maximum statin plus ezetimibe; FOURIER and ODYSSEY OUTCOMES trials proved their efficacy in reducing cardiovascular events in very high-risk patients.
Lifestyle Changes That Reduce Cholesterol and Heart Attack Risk
Evidence-based lifestyle modifications reduce cardiovascular risk through multiple mechanisms simultaneously — addressing not just LDL but inflammation, endothelial function, blood pressure, insulin sensitivity, and body weight.
Dietary modification. A Mediterranean-style dietary pattern — emphasizing vegetables, legumes, whole grains, fish, nuts, and olive oil while limiting saturated fat, processed meat, and refined carbohydrates — reduced major cardiovascular events by approximately 30 percent in the PREDIMED trial. Replacing 1 percent of calories from saturated fat with unsaturated fat reduces LDL-C by approximately 2 mg/dL. Increasing soluble fiber (oats, legumes, psyllium) binds bile acids and reduces hepatic cholesterol recycling.
Physical activity. At least 150 minutes per week of moderate-intensity aerobic exercise improves endothelial function, raises HDL, reduces triglycerides, lowers blood pressure, improves insulin sensitivity, and modestly reduces LDL. The cardiovascular benefit of regular exercise operates through these multiple pathways simultaneously.
Smoking cessation. For smokers, stopping smoking is the single most impactful modifiable risk factor intervention for cardiovascular risk. Within one to two years of cessation, cardiovascular event risk drops significantly; within 10 to 15 years, the excess risk attributable to prior smoking is nearly normalized.
Weight management. Excess body weight — particularly abdominal obesity — drives atherogenic dyslipidemia (elevated triglycerides, low HDL, small dense LDL) and raises LDL through increased hepatic cholesterol synthesis. Weight loss of 5 to 10 percent of body weight consistently improves this lipid pattern. Each kilogram of weight lost is associated with approximately 0.8 mg/dL reduction in LDL-C on average, with larger effects on triglycerides and HDL.
For a foundation on how cholesterol works in the body, see what is cholesterol. To understand how atherosclerosis develops as the disease bridge between cholesterol and heart attack, see what is atherosclerosis. To learn about cholesterol testing, see what is a lipid panel.
Sources: American Heart Association — Heart Attack (heart.org) | Centers for Disease Control and Prevention — Heart Disease Facts (cdc.gov) | National Heart, Lung, and Blood Institute — High Blood Cholesterol (nhlbi.nih.gov) | Cholesterol Treatment Trialists’ Collaboration. Lancet 2010;376:1670–81 | Cannon CP et al. (IMPROVE-IT). NEJM 2015;372:2387–97 | Sabatine MS et al. (FOURIER). NEJM 2017;376:1713–22 | Estruch R et al. (PREDIMED). NEJM 2013/2018
The Cumulative Cholesterol Burden: Why Lifetime Exposure Matters More Than a Single Test
A single cholesterol test captures a snapshot. What actually determines cardiovascular risk is the cumulative exposure of your arteries to atherogenic particles over your lifetime — the total number of LDL particles multiplied by the number of years they have been circulating at elevated concentrations.
This concept — sometimes called “cholesterol years,” analogous to pack-years for tobacco — explains several clinical observations that confuse patients:
Why does a 50-year-old with LDL of 155 mg/dL sometimes have more coronary plaque than a 65-year-old with the same LDL? Because the 50-year-old may have had LDL of 185 mg/dL throughout their twenties and thirties — accumulating plaque rapidly during decades that were not captured by any cholesterol measurement. The 65-year-old may have had lower LDL for decades before it rose to 155 mg/dL in later life, benefiting from lower cumulative exposure.
Why do people with familial hypercholesterolemia — who have LDL of 190 mg/dL or above from birth due to a genetic defect in LDL receptor function — have heart attacks in their thirties and forties, while their peers with the same LDL acquired through diet and sedentary lifestyle may not have events until their fifties or sixties? The answer is decades of additional LDL exposure starting from birth rather than early middle age.
Why do Mendelian randomization studies show that lifelong genetic reduction in LDL of 1 mg/dL reduces cardiovascular risk by approximately 2 percent — roughly twice the benefit of a 1 mg/dL LDL reduction achieved by starting a statin at age 50? Because the genetic reduction applies from birth, while the statin reduction applies only from the point of treatment initiation. Decades of lower cumulative exposure produce proportionally greater protection.
The practical implication of this concept: the earlier cholesterol management begins, the greater the total risk reduction. A person who starts cholesterol-lowering treatment at age 40 instead of 60 does not just get 20 more years of medication — they get 20 more years of slower plaque accumulation, 20 more years of plaque stabilization, and 20 more years of reduced vulnerability to the acute rupture events that cause heart attacks. This is why current guidelines increasingly support earlier evaluation and earlier intervention in younger adults who have significant risk factors, rather than waiting for calculated 10-year risk to cross a threshold that only becomes high enough to act on as patients get older.
Secondary Prevention: Reducing Risk After a Heart Attack
For people who have already had a heart attack, the goal of cholesterol management shifts from prevention of a first event to prevention of a second — because second events carry very high probability without aggressive treatment and are strongly preventable with the right interventions.
After an acute MI, patients are at extremely high near-term cardiovascular risk. The presence of established coronary atherosclerosis demonstrates that the disease process has been underway long enough to produce a vulnerable plaque that ruptured; other plaques almost certainly exist throughout the coronary tree. Without aggressive treatment, approximately 20 percent of patients who survive a first MI will have another major cardiovascular event within five years.
Standard post-MI cholesterol management includes:
High-intensity statin therapy initiated in the hospital, targeting LDL reduction of 50 percent or more from baseline. High-intensity statins (rosuvastatin 20–40 mg or atorvastatin 40–80 mg) are the backbone of secondary prevention. They reduce LDL dramatically, stabilize remaining vulnerable plaques through anti-inflammatory effects, and have been shown in multiple large trials to reduce cardiovascular events by approximately 25 to 35 percent compared to placebo or lower-intensity treatment.
LDL target: below 70 mg/dL, with a goal of below 55 mg/dL in very high-risk patients (those with multiple prior events, concurrent diabetes, CKD, or residual high plaque burden). If LDL remains above 70 mg/dL on maximum-tolerated statin, ezetimibe is added. If still above target, a PCSK9 inhibitor is added. The ODYSSEY OUTCOMES trial demonstrated that alirocumab added to statin therapy reduced major adverse cardiovascular events by 15 percent and reduced mortality in post-ACS patients over a median follow-up of 2.8 years.
Antiplatelet therapy is the other major pharmacological pillar of secondary prevention — reducing the risk of a new thrombus forming at the site of an eroded or ruptured plaque. Aspirin combined with a P2Y12 inhibitor (ticagrelor or clopidogrel) — dual antiplatelet therapy (DAPT) — is standard for 12 months after MI in most patients who have received a stent. Aspirin alone continues indefinitely. The combination of antiplatelet therapy and aggressive LDL lowering addresses both the plaque substrate (LDL-driven atherosclerosis) and the acute thrombotic mechanism (platelet aggregation at the rupture site).
Other secondary prevention medications — ACE inhibitors or ARBs (for patients with reduced ejection fraction or hypertension), beta-blockers (for patients with reduced ejection fraction or ongoing angina), and aldosterone antagonists (for selected patients) — address cardiac remodeling and recurrent ischemia risk through mechanisms complementary to cholesterol management.
Aggressive secondary prevention after a heart attack is the clinical context in which cholesterol management has its largest absolute risk reduction impact. A patient with LDL of 110 mg/dL post-MI who reduces to 55 mg/dL through statin + ezetimibe + PCSK9 inhibitor has a dramatically different 5-year event trajectory than one who receives no treatment or only modest statin therapy.