Cardiomyopathy: A Simple Guide
Cardiomyopathy is a disease of the heart muscle (the myocardium) that impairs the heart’s structural integrity, contractile function, or relaxation properties — independent of coronary artery disease, hypertension, or valvular disease, which are the major external causes of heart failure. In cardiomyopathy, the problem originates within the heart muscle itself: a genetic mutation that produces abnormal sarcomere proteins, a viral infection that triggers destructive autoimmune myocardial inflammation, a toxic exposure that directly poisons cardiomyocytes, or an abnormal protein that infiltrates and stiffens the myocardium. The result in all cases is a heart that cannot function normally — one that is too weak, too stiff, too thick, or too disorganized to pump blood effectively.
Cardiomyopathy is not rare. Hypertrophic cardiomyopathy affects approximately 1 in 500 people — making it one of the most common inherited cardiac conditions — and is the most frequent cause of sudden cardiac death in young athletes. Dilated cardiomyopathy has an estimated prevalence of 1 in 250 and is the most common reason for heart transplantation in the United States. Cardiac amyloidosis, historically considered rare, is now recognized as substantially underdiagnosed — particularly transthyretin amyloid cardiomyopathy (ATTR-CM) in older adults, where it contributes to HFpEF, aortic stenosis, and carpal tunnel syndrome in a recognizable clinical syndrome. Understanding cardiomyopathy — its types, causes, symptoms, genetic implications, and treatment — is increasingly important as diagnostic capabilities improve and therapeutic options expand.
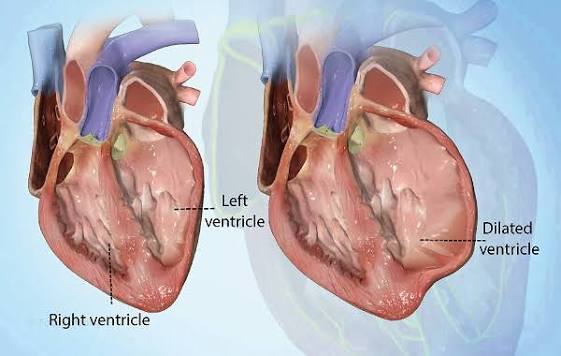
Dilated Cardiomyopathy — An Enlarged, Weakened Heart
Dilated cardiomyopathy (DCM) is characterized by enlargement (dilation) of the left ventricle — and often all four cardiac chambers — accompanied by reduced systolic function (reduced ejection fraction, typically below 40 percent). The dilated ventricle becomes spherical rather than elliptical, reducing the mechanical efficiency of contraction. The thinned, stretched ventricular walls cannot generate adequate contractile force, producing the clinical syndrome of heart failure with reduced ejection fraction (HFrEF).
Dilated cardiomyopathy has numerous causes:
- Idiopathic — no identifiable cause found after thorough evaluation; the most common diagnosis historically, though increasingly genetic testing reveals hereditary mutations in what were previously called “idiopathic” cases
- Genetic/familial — mutations in genes encoding cytoskeletal proteins (titin/TTN is the most common, accounting for 15 to 25 percent of familial DCM), sarcomere proteins (MYH7, MYBPC3), nuclear envelope proteins (lamin A/C, associated with conduction disease and high arrhythmia risk), and others. Familial DCM is autosomal dominant in most cases, meaning first-degree relatives have a 50 percent chance of inheriting the causative variant.
- Viral/post-myocarditis — viral myocarditis (caused by enteroviruses including coxsackievirus B, parvovirus B19, adenovirus, and others) causes acute myocardial inflammation that may resolve, persist as chronic myocarditis, or lead to DCM through immune-mediated ongoing cardiomyocyte damage
- Toxic — alcohol (the most common toxic cause; alcoholic cardiomyopathy may partially recover with sustained abstinence), anthracycline chemotherapy (doxorubicin, epirubicin; dose-dependent, partly preventable with cardioprotective strategies), cocaine, methamphetamine, targeted cancer therapies (trastuzumab, immune checkpoint inhibitors), and others
- Peripartum cardiomyopathy (PPCM) — DCM developing in the final month of pregnancy or within 5 months postpartum, a distinct entity with potential for partial or complete LV function recovery
- Thyroid disease — both hyperthyroidism (high-output heart failure, tachycardia-mediated cardiomyopathy) and severe hypothyroidism can cause reversible DCM
- Tachycardia-induced cardiomyopathy — sustained rapid heart rates from any cause (atrial fibrillation, inappropriate sinus tachycardia, incessant supraventricular tachycardia) can cause DCM that largely or fully recovers when rate control is achieved
Identifying the cause of DCM is clinically important because some causes are reversible (alcohol cessation, rate control, hormone replacement), some require specific treatment modifications (lamin A/C mutations carry high risk of sudden death and require early ICD implantation), and genetic causes mandate family screening.
Hypertrophic Cardiomyopathy — A Thick, Stiff Heart with Obstruction Risk
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiomyopathy, affecting approximately 1 in 500 people across all ethnic groups, caused by mutations in genes encoding sarcomere proteins — the molecular machinery of cardiac muscle contraction. The characteristic finding is asymmetric hypertrophy of the interventricular septum (the wall between the left and right ventricles), which thickens disproportionately relative to the free wall. In approximately two-thirds of patients, this septal hypertrophy causes left ventricular outflow tract (LVOT) obstruction — the thickened septum and the anterior mitral valve leaflet (drawn toward the septum by Venturi effect during systole, a motion called systolic anterior motion or SAM) narrow the outflow tract through which blood exits to the aorta.
HCM symptoms arise from three primary mechanisms: diastolic dysfunction (the thick, stiff ventricle fills poorly under elevated pressures, producing dyspnea), LVOT obstruction (increased afterload limiting cardiac output with exertion), and arrhythmias (the disorganized myocardial architecture predisposes to ventricular arrhythmias and sudden cardiac death, and the left atrial hypertension and enlargement predispose to atrial fibrillation). Common presenting symptoms include exertional dyspnea and reduced exercise tolerance, chest pain (from microvascular ischemia even without coronary artery disease), palpitations, and — in a minority of patients — syncope or presyncope, which warrants urgent evaluation for LVOT obstruction severity and sudden death risk.
Restrictive Cardiomyopathy — Stiff Ventricles That Cannot Fill
Restrictive cardiomyopathy is the least common of the three primary cardiomyopathy types, characterized by abnormal diastolic function — the ventricular walls are stiff and non-compliant, severely restricting ventricular filling during diastole — with preserved or near-normal systolic function (normal or reduced but not severely impaired ejection fraction). The physiological consequence is markedly elevated ventricular filling pressures with relatively small ventricular volumes: the heart cannot fill adequately despite high filling pressures, limiting stroke volume and cardiac output. Biatrial enlargement is characteristic, as chronically elevated ventricular filling pressures back up into the atria, producing the “four-chamber enlargement” pattern with small, non-dilated ventricles — distinguishing restrictive physiology from dilated cardiomyopathy echocardiographically.
Restrictive cardiomyopathy is most commonly caused by infiltrative diseases that deposit abnormal material within the myocardium:
Cardiac amyloidosis is the most clinically important cause of restrictive cardiomyopathy. Amyloid fibrils — abnormal protein aggregates with characteristic beta-sheet structure — deposit in the myocardium, stiffening the ventricular walls, reducing diastolic compliance, and producing a restrictive pattern. Two forms predominate clinically: AL (light chain) amyloidosis, caused by a plasma cell dyscrasia that produces amyloidogenic immunoglobulin light chains (associated with multiple myeloma and related conditions, requiring chemotherapy directed at the underlying plasma cell clone), and transthyretin amyloid cardiomyopathy (ATTR-CM), in which the liver-produced transthyretin protein misfolds and aggregates in the myocardium. ATTR-CM occurs in two forms: wild-type (acquired, affecting primarily older men, formerly called “senile” amyloidosis) and hereditary (caused by TTR gene mutations, more common in Black Americans due to the prevalent Val122Ile variant). Tafamidis, a TTR stabilizer approved in 2019, significantly reduces cardiovascular mortality and hospitalization in ATTR-CM and represents the first disease-modifying therapy for this previously untreatable condition.
Cardiac sarcoidosis deposits granulomatous inflammation in the myocardium, producing patchy fibrosis and necrosis that can cause both restrictive physiology and, critically, ventricular arrhythmias and high-degree heart block. Cardiac sarcoidosis is an important treatable cause of unexplained ventricular arrhythmias, complete heart block in younger patients, and cardiomyopathy — immunosuppressive therapy with corticosteroids can reduce active inflammation, and ICD implantation is important for patients with significant arrhythmia risk.
Genetics, Family Screening, and Sudden Death Risk
Cardiomyopathy genetics is a rapidly evolving field with important clinical implications for patients and their families. Many cardiomyopathies — particularly HCM, DCM, and arrhythmogenic cardiomyopathy — are inherited as autosomal dominant conditions, meaning a pathogenic variant in one copy of the relevant gene is sufficient to cause disease (though with variable penetrance — not all variant carriers develop clinical cardiomyopathy).
Genetic testing in a proband (the first identified affected family member) can identify the causative variant in 40 to 60 percent of HCM patients (most commonly in MYH7 or MYBPC3) and in 15 to 35 percent of DCM patients (titin/TTN most common, plus LMNA, MYH7, SCN5A, and others). Identifying the causative variant enables cascade screening — testing first-degree relatives (parents, siblings, children) for the same variant. Relatives who test positive for the variant require clinical evaluation and ongoing cardiac surveillance even if currently asymptomatic, since cardiomyopathy may be absent or mild in youth and progressively penetrant with aging. Relatives who test negative have dramatically reduced disease risk and typically do not require ongoing cardiac surveillance beyond standard preventive care.
Sudden cardiac death (SCD) risk stratification is a critical component of cardiomyopathy management for both HCM and ARVC. In HCM, SCD risk factors include prior cardiac arrest or sustained VT, massive LV hypertrophy (wall thickness 30 mm or above), unexplained syncope, non-sustained VT on monitoring, family history of HCM-related SCD, and LV apical aneurysm. The 2020 AHA/ACC HCM guidelines provide a recommended SCD risk calculator that incorporates these factors, and patients at intermediate or high 5-year SCD risk (2.4 percent or higher) are generally offered prophylactic ICD implantation. In ARVC, major risk factors for SCD include prior cardiac arrest, sustained VT, frequent PVCs, right ventricular dysfunction severity, and competitive athletic participation — which dramatically worsens arrhythmia risk in ARVC patients and is a strong indication for athletic restriction.
The American Heart Association’s cardiomyopathy resources provide comprehensive patient education on all cardiomyopathy types. The CDC cardiomyopathy overview covers types, causes, and management basics. The NHLBI cardiomyopathy guide addresses symptoms, diagnosis, treatment options, and living with cardiomyopathy.
Related reading: Heart Failure | Coronary Artery Disease | What Causes Heart Disease? | Major Risk Factors for Heart Disease | Heart Attack Prevention
Sources
- Ommen SR, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2020;76(25):e159-e240.
- Heidenreich PA, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure. J Am Coll Cardiol. 2022;79(17):e263-e421.
- Rapezzi C, et al. Transthyretin-Related Amyloidoses and the Heart: A Clinical Overview. Nat Rev Cardiol. 2010;7(7):398-408.
- Schultheiss HP, et al. Dilated Cardiomyopathy. Nat Rev Dis Primers. 2019;5(1):32.
- Corrado D, et al. Arrhythmogenic Cardiomyopathy: Definition, Diagnosis, and Treatment. Eur Heart J. 2023;44(34):3320-3348.
- Maurer MS, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-ACT). N Engl J Med. 2018;379(11):1007-1016.
Arrhythmogenic Cardiomyopathy — When Muscle Turns to Fat and Fibrosis
Arrhythmogenic cardiomyopathy (ACM) — previously called arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) — is an inherited cardiomyopathy caused by mutations in genes encoding desmosomal proteins, the structural molecules that hold cardiomyocytes together and maintain myocardial integrity. Pathogenic variants in PKP2 (plakophilin-2), DSP (desmoplakin), DSC2, DSG2, and other desmosomal genes disrupt cell-cell adhesion in the myocardium, causing cardiomyocyte death under mechanical stress that is replaced by fibrofatty tissue. This fibrofatty replacement creates the structural substrate for ventricular arrhythmias — the fibrosis creates channels of slow conduction that support reentrant circuits producing ventricular tachycardia.
ACM is a primary cause of sudden cardiac death in young people and athletes, and a leading indication for ICD implantation in patients under 35 years of age. The condition predominantly affects the right ventricle in its classic form, producing right ventricular dilation, regional wall motion abnormalities, and RV functional impairment on echocardiography and cardiac MRI. Left-dominant ACM — where left ventricular involvement predominates — is increasingly recognized, particularly in patients with DSP and FLNC mutations, and can masquerade as dilated cardiomyopathy or be misclassified if standard ARVC diagnostic criteria focused on the RV are applied without LV assessment.
Athletic exercise dramatically worsens arrhythmia risk in ACM by increasing mechanical stress on the diseased desmosomal connections, accelerating fibrofatty replacement, and provoking arrhythmias. Competitive athletic restriction is one of the most consistently recommended and emotionally difficult interventions in ACM management — patients who are young, athletic, and symptom-free often struggle to accept restriction from the sport that may have first brought the diagnosis to attention. The 2019 HRS Expert Consensus on ACM recommends athletic restriction for all patients with ACM, including those without clinical disease expression, due to the catastrophic consequences of exercise-triggered SCD in a condition where the substrate for fatal arrhythmia may exist before echocardiographic or MRI abnormalities are apparent.
Treatment Approaches by Cardiomyopathy Type
Treatment of cardiomyopathy depends critically on type — the pathophysiological mechanisms differ substantially across subtypes, making the treatment for one type potentially harmful in another. This is one reason accurate diagnosis and subtype classification matters clinically, not merely academically.
Dilated cardiomyopathy treatment follows guideline-directed medical therapy (GDMT) for heart failure with reduced ejection fraction: the four-drug combination of ARNI/ACE inhibitor/ARB plus beta-blocker plus mineralocorticoid receptor antagonist plus SGLT2 inhibitor has demonstrated combined mortality reductions of 50 to 60 percent compared to placebo in HFrEF trials. Device therapy — cardiac resynchronization therapy (CRT) for patients with left bundle branch block and QRS duration above 150 ms, and ICD for primary prevention of sudden death in patients with LVEF persistently below 35 percent on optimal medical therapy — adds incremental survival benefit. Specific DCM causes require additional directed therapy: alcohol abstinence for alcoholic cardiomyopathy, rate control and rhythm restoration for tachycardia-induced cardiomyopathy, chemotherapy modification or cardioprotection for anthracycline cardiomyopathy, and immunosuppression for giant cell myocarditis or cardiac sarcoidosis.
Hypertrophic cardiomyopathy treatment targets the specific pathophysiological abnormalities: diastolic dysfunction, LVOT obstruction, and arrhythmia risk. Beta-blockers are the mainstay of symptom management in obstructive HCM, reducing heart rate (which prolongs diastolic filling time and reduces the dynamic LVOT gradient), decreasing myocardial oxygen demand, and providing some antiarrhythmic effect. Non-dihydropyridine calcium channel blockers (verapamil, diltiazem) are an alternative in patients who cannot tolerate beta-blockers. Disopyramide, a class Ia antiarrhythmic with negative inotropic properties, can reduce LVOT gradient when added to beta-blocker therapy in patients with refractory obstructive symptoms.
A transformative therapeutic advance in HCM is mavacamten, a selective cardiac myosin inhibitor approved by the FDA in 2022 for symptomatic obstructive HCM. Mavacamten directly addresses the pathophysiology of HCM by inhibiting the hypercontractile myosin motor proteins that cause excessive force generation and LVOT obstruction. In the EXPLORER-HCM trial, mavacamten significantly reduced LVOT gradient, improved NYHA functional class, and improved exercise capacity compared to placebo — representing the first disease-mechanism-targeted pharmacotherapy for HCM. Patients who remain severely symptomatic despite medical therapy may be candidates for invasive septal reduction: surgical myectomy (the gold standard at expert centers, removing obstructing septal muscle) or alcohol septal ablation (catheter-delivered alcohol that selectively destroys a portion of the interventricular septum to reduce obstruction).
Restrictive cardiomyopathy and amyloidosis treatment has historically been limited to symptom management (diuretics for congestion, pacemakers for heart block) without disease-modifying options. The approval of tafamidis for ATTR-CM in 2019 changed this for transthyretin amyloidosis — the TTR-ACT trial demonstrated 30 percent reduction in cardiovascular mortality and 32 percent reduction in cardiovascular hospitalization over 30 months. Newer agents in clinical development include CRISPR-based TTR gene editing (which virtually eliminates hepatic TTR production in early trials), RNA interference agents (vutrisiran, already approved for hereditary TTR polyneuropathy), and small molecule disruptors. For AL amyloidosis, early and effective treatment of the underlying plasma cell dyscrasia with chemotherapy and autologous stem cell transplantation (in eligible patients) can halt amyloid production and in some cases allow partial myocardial recovery.
Diagnosing Cardiomyopathy — From Echocardiography to Genetic Panel
Cardiomyopathy evaluation is a systematic process that typically proceeds from clinical suspicion to echocardiography to cardiac MRI to genetic testing, with the specific pathway determined by clinical presentation, echocardiographic findings, and suspected etiology.
Echocardiography is the first-line imaging modality, providing measurement of LV dimensions, wall thickness, ejection fraction, and diastolic parameters that suggest the cardiomyopathy type: dilated LV with thin walls and reduced LVEF in DCM; asymmetric septal hypertrophy with possible LVOT obstruction in HCM; small non-dilated ventricles with biatrial enlargement and restrictive filling in restrictive/infiltrative cardiomyopathy; RV dilation and regional wall motion abnormalities in ACM. Speckle tracking echocardiography — which measures global longitudinal strain (GLS), a more sensitive index of myocardial function than LVEF — can detect subclinical myocardial dysfunction before LVEF falls, useful for surveillance of cardiomyopathy gene carriers and cardiotoxicity monitoring.
Cardiac MRI (CMR) is increasingly essential in cardiomyopathy evaluation, providing highly accurate ventricular volume and function measurement, detailed myocardial tissue characterization through late gadolinium enhancement (LGE) imaging and T1/T2 mapping, and pattern recognition that helps identify specific etiologies. The LGE pattern is particularly informative: subendocardial or transmural LGE in a coronary distribution indicates ischemic cardiomyopathy; mid-wall LGE in a non-coronary distribution suggests idiopathic or genetic DCM; patchy or diffuse LGE in HCM correlates with fibrosis extent and SCD risk; “ground glass” subendocardial LGE with native T1 elevation and low native T1 in fat-replaced areas in ACM; and diffuse subendocardial LGE with T1 elevation and ECV (extracellular volume) expansion is characteristic of amyloidosis.
Genetic testing with comprehensive cardiomyopathy panel sequencing (covering 50 to 100 genes relevant to inherited cardiomyopathies) is now recommended for most patients with cardiomyopathy to identify pathogenic variants, guide family screening, and in some cases refine prognosis (e.g., LMNA mutations in DCM carry high risk of malignant arrhythmias and often require earlier ICD implantation). Genetic counseling — by specialists trained in cardiovascular genetics — is an essential companion to genetic testing, helping patients and families understand the implications of positive, negative, and variant-of-uncertain-significance results.
Living With Cardiomyopathy — Long-Term Perspective
Cardiomyopathy is a lifelong condition in most cases, but the prognosis varies enormously by type, severity at diagnosis, response to treatment, and access to expert care. Many patients with dilated cardiomyopathy experience partial or complete recovery of LV function with optimal medical therapy — particularly younger patients, those with identified reversible causes (tachycardia-mediated, peripartum, toxic, viral), and those who receive early, aggressive GDMT. HCM patients who receive appropriate management (symptoms addressed, SCD risk stratified and mitigated with ICD when indicated) have life expectancy approaching the general population — a striking change from the historical perception of HCM as a highly lethal condition. ATTR-CM patients treated with tafamidis have meaningfully improved survival and quality of life outcomes compared to the untreated natural history.
Lifestyle considerations in cardiomyopathy include: moderate aerobic exercise (beneficial for HFrEF patients and acceptable in HCM patients without significant obstruction or high arrhythmia risk); sodium restriction and fluid management (particularly important when heart failure is present); avoidance of cardiotoxic substances (alcohol in all cardiomyopathy patients, recreational drugs); medication adherence (as important in cardiomyopathy as in coronary disease); and — critically in HCM and ACM — evaluation by a multidisciplinary team experienced in these conditions before any decisions about athletic participation or reproductive planning. Cardiomyopathy management benefits greatly from care at centers with dedicated cardiomyopathy programs and hereditary heart disease expertise, where the full spectrum of evaluation (genetic testing, advanced imaging, electrophysiology, cardiac surgery) and treatment (medical, device, and interventional) is available.