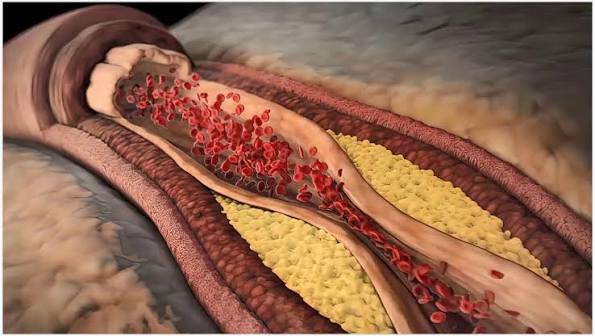
Most people picture cholesterol and plaque buildup in arteries the way they picture scale inside a water pipe — fat coating the inside of a tube until it narrows enough to slow or stop flow. That picture is wrong in important ways, and the difference matters for how you think about risk, treatment, and what’s actually happening in your body.
Atherosclerosis — the medical term for the disease that produces arterial plaque — is not a passive accumulation of grease. It is a chronic inflammatory process that unfolds over decades, embedded within the wall of arteries rather than coating the surface. It begins before most people are old enough to think about their heart. It is usually silent until it is not — and when it is not, the consequences include heart attack, stroke, and peripheral artery disease.
Understanding how cholesterol and plaque form in arteries changes how you interpret your cholesterol numbers, what kinds of risk reduction actually matter, and why standard screening detects some cardiovascular risks while missing others. This article explains the process from the first cellular events through to clinical intervention.
What Plaque Is and Where It Forms
Atherosclerotic plaque is a deposit of lipids, immune cells, smooth muscle cells, extracellular matrix, and eventually calcium within the inner wall of an artery. The key word is within — plaque develops between layers of the arterial wall, not as a layer sitting on top of the inner surface.
Arteries have three layers: the intima (inner lining of endothelial cells), the media (smooth muscle layer), and the adventitia (outer connective tissue). Plaque forms in the intima, and as it grows it can expand outward into the media or inward (toward the arterial lumen), progressively narrowing the channel through which blood flows.
Plaque does not form uniformly throughout the circulatory system. It develops preferentially at locations experiencing turbulent or disturbed blood flow: branch points, curves, and bifurcations. These are the places where mechanical stress on the endothelium is highest, and where the initial injury that starts the process is most likely to occur.
The most clinically important locations are:
- Coronary arteries — plaques here cause coronary artery disease (CAD); when a plaque ruptures or a vessel narrows severely, the result is myocardial infarction
- Carotid arteries — plaque at the carotid bifurcation in the neck is the most common source of emboli that cause ischemic stroke
- Peripheral arteries (iliac, femoral, popliteal) — produce peripheral artery disease (PAD), causing pain with walking (claudication) and, in severe cases, limb ischemia
- Aorta — contributes to abdominal aortic aneurysm risk and can be a source of emboli
- Renal arteries — plaque-related stenosis raises blood pressure and damages kidney function
Because atherosclerosis is driven by systemic risk factors — LDL, blood pressure, smoking, diabetes — it is a systemic disease. Significant plaque at one site almost always means plaque at multiple sites, even if only one location is causing symptoms.
Cholesterol and Plaque Buildup in Arteries — The Step-by-Step Process
The development of an atherosclerotic plaque follows a recognizable sequence. Each step makes the next more likely, and the process can be interrupted at multiple points by reducing the underlying risk factors.
Step 1: Endothelial injury. The endothelium — the single layer of cells lining the inside of arteries — is not a passive barrier. It regulates vascular tone, controls permeability, and actively prevents the adhesion of immune cells and platelets. When endothelial cells are damaged or dysfunctional, this protective function breaks down. The triggers for endothelial injury include chronic hypertension (mechanical shear stress), cigarette smoke (oxidative stress and direct toxicity), elevated blood glucose (glycation of endothelial proteins), and the presence of oxidized LDL itself. Once injured, endothelial cells upregulate adhesion molecules — VCAM-1 and ICAM-1 — that signal immune cells to adhere and enter.
Step 2: LDL infiltration and oxidation. When the endothelium becomes more permeable, LDL particles cross into the intima more readily. Once in the intima, LDL particles encounter an oxidative environment and are modified — most significantly, oxidized. Oxidized LDL (oxLDL) is qualitatively different from native LDL: it is recognized by specific receptors on macrophages, it directly stimulates inflammation, and it is toxic to endothelial cells, creating a reinforcing cycle of injury and dysfunction.
Step 3: Monocyte recruitment and macrophage differentiation. Endothelial adhesion molecules capture circulating monocytes, which then transmigrate across the endothelium into the intima. There they differentiate into macrophages — the primary cleanup cells of the immune system. The macrophages’ job, in theory, is to engulf and process the oxidized LDL, clearing it from the vessel wall.
Step 4: Foam cell formation. Macrophages engulf oxidized LDL through scavenger receptors (SR-A, CD36). Unlike the standard LDL receptor pathway — which is feedback-regulated so that cells stop taking up LDL when they have enough — scavenger receptors are not downregulated by intracellular lipid accumulation. Macrophages continue taking in oxLDL until they are so engorged with lipid droplets that they become “foam cells” — named for their foamy appearance under the microscope. Foam cells are the pathological hallmark of early atherosclerosis.
Step 5: The fatty streak. As foam cells accumulate, they form the fatty streak — a yellow, slightly raised deposit visible to the naked eye on the inner surface of arteries. Fatty streaks are found in the coronary arteries and aorta of virtually all children in Western societies by age 10 and are nearly universal by age 20 in American autopsy series. They are not yet flow-limiting or clinically significant, but they are the direct precursor to established plaque.
Step 6: Smooth muscle cell migration and matrix accumulation. Growth factors released by foam cells and other inflammatory cells stimulate smooth muscle cells to migrate from the media (the muscular layer of the artery) into the intima. There, smooth muscle cells begin secreting collagen and other extracellular matrix proteins, creating a growing structural scaffold around the lipid-laden core. This accumulation of smooth muscle cells and connective tissue is what transforms the fatty streak into a more complex, growing plaque.
Step 7: Formation of the fibrous plaque. A mature atherosclerotic plaque consists of a necrotic lipid-rich core — containing dead foam cells, cholesterol crystals, extracellular lipid, and inflammatory cells — covered by a fibrous cap composed of smooth muscle cells embedded in collagen. The cap physically separates the thrombogenic material in the core from the flowing blood. As long as the cap remains intact, the plaque may cause gradual narrowing but usually does not trigger an acute event.
Why Some Plaques Are Dangerous and Others Aren’t
Not all plaques are equally dangerous — and the distinction is not simply about how much the plaque has narrowed the artery.
Stable plaque has a thick, collagen-rich fibrous cap, a relatively small necrotic lipid core, fewer macrophages, and more smooth muscle cells. Stable plaques may calcify significantly over time, making them denser and more mechanically stable. They tend to cause symptoms through gradual narrowing — producing stable angina (predictable chest pain with exertion) — but are less likely to rupture and trigger an acute event.
Vulnerable plaque (technically called thin-cap fibroatheroma) has the opposite profile: a thin fibrous cap (less than 65 micrometers thick), a large necrotic lipid core, heavy infiltration by macrophages and T cells, and few smooth muscle cells. The macrophages in vulnerable plaques release matrix metalloproteinases (MMPs) that degrade collagen, weakening the cap. The thin, inflamed cap over a large lipid lake is mechanically unstable and prone to rupture.
When a vulnerable plaque ruptures, the necrotic core — rich in tissue factor and collagen — is exposed to flowing blood. Platelets rush in, the coagulation cascade activates, and a thrombus (blood clot) forms rapidly. If the thrombus occludes the artery completely, downstream tissue dies: the result is a heart attack (in the coronary arteries) or stroke (in the carotid or cerebral arteries).
This leads to one of the most clinically counterintuitive facts in cardiology: the plaques most likely to cause a heart attack are not necessarily the most obstructive. Large, calcified, stable plaques may narrow an artery by 70 or 80 percent but remain stable for years. A smaller, softer, vulnerable plaque that narrows the artery by only 40 or 50 percent may rupture first and cause a fatal MI. This is why imaging that measures stenosis (how blocked the artery is) does not fully capture rupture risk, and why risk-reduction strategies targeting the inflammatory and lipid-driven processes that create vulnerable plaques are at least as important as treating symptoms of stable disease.
The Role of LDL Particles in Plaque Formation
Not all LDL is identical, and understanding what LDL particles actually do in the artery wall helps explain why particle number and size matter, not just total LDL cholesterol concentration.
LDL provides the raw material. Foam cells form from macrophages engulfing oxidized LDL. More LDL particles crossing the endothelium means more substrate for foam cell formation. This is why lowering LDL — even from levels previously considered “normal” — reduces cardiovascular events: less substrate means less foam cell formation and slower plaque growth.
LDL particle number (LDL-P / ApoB) predicts plaque better than LDL-C. LDL-C measures the amount of cholesterol carried in LDL particles. LDL-P (or its proxy ApoB, since each LDL particle contains one ApoB molecule) measures the number of LDL particles. Two people can have the same LDL-C but very different LDL-P if their particles are different sizes. Research consistently shows that LDL-P and ApoB predict cardiovascular events more accurately than LDL-C alone.
Small, dense LDL is more atherogenic. Small, dense LDL particles penetrate the endothelium more easily, are retained in the arterial wall longer, and are more susceptible to oxidation than large, buoyant LDL particles. Triglyceride-rich states (metabolic syndrome, diabetes, high-carbohydrate diets) tend to produce more small dense LDL — which is one mechanism by which high triglycerides and insulin resistance increase cardiovascular risk beyond their direct effects.
Lp(a) is a particularly potent atherogenic particle. Lipoprotein(a) — Lp(a) — is a modified form of LDL with an additional protein (apo(a)) attached. Lp(a) levels are almost entirely genetically determined, unresponsive to most dietary changes, and resistant to most currently available lipid-lowering medications. Lp(a) promotes atherosclerosis through multiple mechanisms: it accumulates in plaque, carries oxidized phospholipids, inhibits fibrinolysis, and promotes thrombosis after plaque rupture.
HDL and reverse cholesterol transport. HDL particles participate in reverse cholesterol transport — they accept cholesterol from peripheral cells, including foam cells in plaques, and shuttle it back to the liver for excretion or recycling. This is the mechanism by which HDL is thought to be protective. However, raising HDL pharmacologically has not consistently reduced cardiovascular events, suggesting that HDL functionality — not just HDL-C concentration — is the relevant factor.
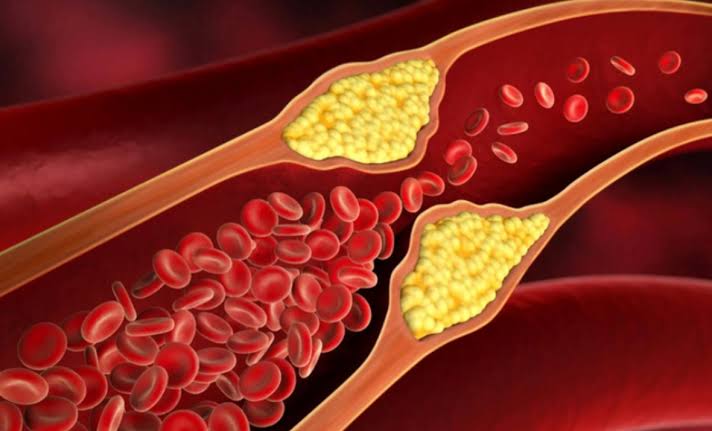
How Fast Does Plaque Develop?
The timeline of atherosclerosis is one of the most sobering facts in medicine. The disease does not begin at age 50 when someone gets their first cholesterol test.
Autopsy studies of American men and women who died of non-cardiac causes document fatty streaks in the coronary arteries and aorta beginning in the second decade of life — between ages 10 and 20. By the third and fourth decades, fibrous plaques are common. By middle age, significant coronary atherosclerosis is nearly universal in populations with Western dietary and lifestyle patterns.
The most extensive autopsy data on early atherosclerosis come from the PDAY (Pathobiological Determinants of Atherosclerosis in Youth) study, which examined coronary arteries and aortas of 1,532 individuals aged 15 to 34 who died of non-cardiac causes (accidents, homicides, suicides). Raised atherosclerotic lesions were found in the right coronary artery of 2% of 15–19 year-olds and increased steadily with age. Prevalence and extent of lesions correlated strongly with antemortem risk factors: smoking, hypertension, obesity, and elevated non-HDL cholesterol.
The clinical implication: the cholesterol-fueled inflammatory process that ultimately causes a heart attack at age 55 or 65 has typically been running for 30 or 40 years. The decades during which this process is silent are not wasted time for prevention — they are the window in which intervention is most effective.
Plaque progression is not linear. Most of the time it is slow and subclinical. But certain periods or conditions can accelerate it: periods of sustained high LDL, the development of diabetes, a decade of smoking, or a prolonged untreated hypertensive state can each accelerate the rate of plaque accumulation and destabilization.
Can Plaque Be Reversed or Stopped?
This is the question most patients want answered, and the evidence is more nuanced than a simple yes or no.
Full plaque reversal is not achievable with current therapies. Plaque volume cannot be eliminated. But two important things can be achieved: partial regression (modest reduction in plaque volume) and — more importantly — plaque stabilization (transforming vulnerable, rupture-prone plaques into stable, calcified, less dangerous ones).
The most rigorous evidence comes from intravascular ultrasound (IVUS) trials, which directly measure coronary plaque volume in living patients.
The ASTEROID trial randomized patients to high-dose rosuvastatin (40 mg), achieving a mean LDL reduction to 60.8 mg/dL. After 24 months of treatment, IVUS showed statistically significant regression of coronary plaque volume — a landmark result that confirmed intensive lipid lowering could partially reverse established plaque. The REVERSAL trial compared intensive atorvastatin (80 mg) vs. moderate pravastatin (40 mg). Intensive therapy halted plaque progression entirely; moderate therapy produced slow but continued progression. More recent data with PCSK9 inhibitors achieving LDL below 55 mg/dL show greater regression than statin trials alone, suggesting that lower is better for plaque regression too.
Plaque stabilization may matter more than regression. When statins reduce LDL aggressively, plaques that remain often show increased calcification on CT imaging. This might seem alarming, but calcification in this context likely represents plaque stabilization — the necrotic lipid core shrinks, the fibrous cap becomes thicker and more calcified, and the plaque becomes mechanically more resistant to rupture. Multiple large trials confirm that statins dramatically reduce cardiovascular events without producing dramatic changes in plaque volume — suggesting that much of the benefit comes from stabilizing vulnerable plaques rather than shrinking total plaque burden.
Measuring Plaque Without Surgery
Because atherosclerosis is invisible without imaging, several non-invasive tools allow physicians to directly or indirectly estimate plaque burden.
Coronary Artery Calcium Score (CAC). A CT scan of the heart detects calcified plaque within the coronary arteries and quantifies it as an Agatston score. CAC = 0 indicates very low 10-year cardiovascular event risk, appropriate for deferring statin therapy in intermediate-risk patients. CAC 1–99 generally favors initiating statin therapy. CAC 100–299 indicates moderate plaque burden; statin therapy is clearly indicated. CAC ≥ 300 (or ≥ 75th percentile for age/sex/race) indicates high plaque burden requiring high-intensity statin therapy and aggressive risk factor management. Current ACC/AHA guidelines endorse CAC scoring for intermediate-risk adults when the statin treatment decision is uncertain.
Carotid Intima-Media Thickness (CIMT). Ultrasound measurement of the thickness of the carotid artery wall at the intima-media interface. Increased CIMT correlates with cardiovascular risk and is used as a surrogate marker for subclinical atherosclerosis in research and some clinical settings.
CT Coronary Angiography (CTCA). A contrast-enhanced CT scan that visualizes the coronary arteries directly, identifying both plaque and the degree of stenosis. CTCA detects both calcified and non-calcified (soft) plaque — the latter being more concerning for near-term rupture risk. Recent guidelines support CTCA as a diagnostic tool for stable chest pain and for evaluating intermediate-risk patients.
How to Slow Plaque Buildup
Because atherosclerosis is a chronic process driven by modifiable risk factors, every year of effective risk factor control reduces the cumulative plaque burden and the probability of rupture.
Lower LDL aggressively. Meta-analyses of statin trials consistently show that each 1 mmol/L (approximately 39 mg/dL) reduction in LDL-C is associated with roughly a 22 percent relative reduction in major cardiovascular events. More LDL reduction produces more benefit. Current guidelines recommend target LDL below 70 mg/dL for high-risk patients and below 55 mg/dL for very high-risk patients. If statins alone are insufficient, ezetimibe and PCSK9 inhibitors are proven add-on therapies. For more on understanding your LDL numbers, see our guides to LDL vs HDL cholesterol and how to understand your total cholesterol results.
Control blood pressure. Hypertension is one of the primary drivers of endothelial injury — the first step in plaque formation. Blood pressure control to below 130/80 mmHg reduces cardiovascular event rates substantially and slows plaque progression.
Stop smoking. Cigarette smoking accelerates every step of the atherosclerotic process — endothelial injury, LDL oxidation, foam cell formation, plaque inflammation, and rupture risk. The benefits of smoking cessation on cardiovascular risk begin within weeks and are clinically significant within one to two years.
Control blood sugar. In people with diabetes or prediabetes, elevated blood glucose promotes oxidative modification of LDL particles and accelerated endothelial dysfunction. Glycemic control reduces cardiovascular events in people with diabetes.
Adopt a heart-healthy dietary pattern. A Mediterranean-style dietary pattern has been shown in randomized trials (most notably PREDIMED) to reduce major cardiovascular events by approximately 30 percent. The dietary benefit operates through multiple mechanisms: lowering LDL, reducing triglycerides, improving endothelial function, and reducing systemic inflammation.
Exercise regularly. Aerobic exercise improves endothelial function directly and reduces multiple cardiovascular risk factors simultaneously: LDL, triglycerides, blood pressure, insulin resistance, and body weight. Guidelines recommend at least 150 minutes per week of moderate-intensity aerobic activity.
For more on the basics of cholesterol and why it matters, see what is cholesterol. For information on cholesterol testing and what your numbers mean, see high cholesterol symptoms and why testing matters. For the broader cardiovascular disease that plaque buildup causes, see what is atherosclerosis.
Sources: American Heart Association — Atherosclerosis (heart.org) | National Heart, Lung, and Blood Institute — Atherosclerosis (nhlbi.nih.gov) | Centers for Disease Control and Prevention — Heart Disease Facts (cdc.gov) | Nissen SE et al. ASTEROID Trial. JAMA 2006;295:1556–65 | Nissen SE et al. REVERSAL Trial. JAMA 2004;291:1071–80 | McGill HC et al. PDAY Study. Arterioscler Thromb Vasc Biol 2000;20:1998–2004
The Connection Between Plaque, Inflammation, and C-Reactive Protein
Atherosclerosis is fundamentally an inflammatory disease, and this means that markers of systemic inflammation can provide additional risk information beyond the standard lipid panel. The most clinically studied is high-sensitivity C-reactive protein (hs-CRP) — a protein produced by the liver in response to inflammatory signals, including those originating from activated macrophages within arterial plaques.
Elevated hs-CRP (above 2 mg/L, and particularly above 3 mg/L) is associated with increased cardiovascular risk independently of LDL cholesterol. The JUPITER trial demonstrated that patients with low LDL but elevated hs-CRP benefited from statin therapy — reducing both LDL and hs-CRP — with a significant reduction in cardiovascular events. This finding helped establish the concept that inflammation is not merely a byproduct of plaque development but an active contributor to cardiovascular risk that can be targeted therapeutically.
More recent data has come from the CANTOS trial, which tested canakinumab — a targeted anti-inflammatory therapy that inhibits IL-1β, one of the key cytokines driving plaque inflammation — in patients with prior myocardial infarction and elevated hs-CRP. Patients receiving canakinumab had significant reductions in recurrent cardiovascular events compared to placebo, despite no change in LDL. This was the first large trial to demonstrate that directly targeting inflammation (without any lipid-lowering effect) reduces cardiovascular events — confirming the inflammatory hypothesis of atherosclerosis in a prospective, randomized clinical trial.
For patients, this means two things: first, hs-CRP testing can provide additional risk information in cases where the decision to treat is uncertain based on LDL alone. Second, therapies that reduce plaque inflammation — including statins (which have anti-inflammatory properties independent of LDL lowering), aspirin, and emerging targeted therapies — work partly by stabilizing vulnerable plaques through reducing the macrophage-driven inflammation that thins fibrous caps.