Cholesterol and Stroke Risk: What the Numbers Mean

Cholesterol and stroke risk are linked through the pathophysiology of atherosclerosis — the process by which LDL cholesterol-driven plaque accumulates in arterial walls, progressively narrowing the lumen and creating vulnerable surfaces prone to rupture and thrombosis. While the relationship between cholesterol and stroke is somewhat more nuanced than the straightforward association between cholesterol and heart attack risk, high LDL cholesterol is a well-established independent risk factor for ischemic stroke from large artery atherosclerosis and small vessel disease, and statin therapy targeting LDL reduction is a cornerstone of ischemic stroke prevention.
The nuance in the cholesterol-stroke relationship arises partly from the heterogeneity of stroke — not all strokes are caused by atherosclerosis. Cardioembolic stroke (from atrial fibrillation or other cardiac sources) and hemorrhagic stroke (from vessel rupture) are not directly caused by high cholesterol, and the lipid-lowering benefit in stroke prevention is primarily confined to ischemic stroke from atherosclerotic mechanisms. Furthermore, some early epidemiological studies showed a paradoxical relationship between very low total cholesterol and hemorrhagic stroke risk — an observation that reflected confounding by liver disease (which lowers both cholesterol and coagulation factors, increasing bleeding risk) and has not been observed with statin-induced LDL reduction in clinical trials.
For patients with ischemic stroke or TIA, particularly those with evidence of large artery atherosclerosis or carotid stenosis, aggressive LDL lowering with high-intensity statin therapy is among the most evidence-based and impactful secondary prevention interventions available.
How LDL Cholesterol Drives Atherosclerosis and Stroke
The atherogenic process that connects elevated LDL cholesterol to ischemic stroke begins at the level of the arterial endothelium and unfolds over years to decades. Understanding this process clarifies why LDL reduction is so powerfully effective as a stroke prevention strategy:
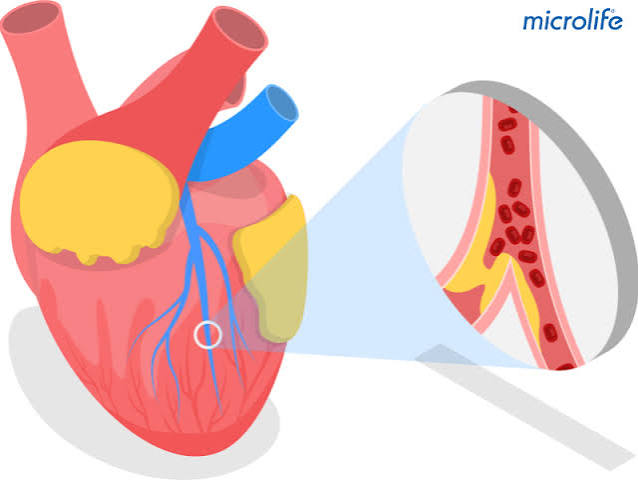
LDL particles — lipoproteins carrying cholesterol through the bloodstream — penetrate the arterial intima (the innermost arterial wall layer) at sites of endothelial dysfunction. Endothelial dysfunction is caused and perpetuated by the same traditional cardiovascular risk factors that drive stroke: hypertension, smoking, diabetes, and elevated LDL itself. At sites of dysfunction, the normally tight endothelial barrier becomes more permeable, allowing LDL to enter the subintimal space.
In the arterial wall, LDL undergoes oxidative modification — becoming oxidized LDL (oxLDL), which is biologically far more dangerous than native LDL. OxLDL is recognized by scavenger receptors on macrophages recruited to the arterial wall. Unlike normal LDL receptor-mediated uptake (which is regulated and downregulated when cholesterol is sufficient), scavenger receptor-mediated uptake of oxLDL is unregulated — macrophages continue engulfing oxLDL until they become lipid-laden “foam cells.” Accumulating foam cells form the fatty streak, the earliest visible lesion of atherosclerosis, which can be detected at autopsy in arteries of children and young adults.
Over years to decades, the fatty streak evolves into an advanced atherosclerotic plaque: the lipid core grows as foam cells die and release their cholesterol; smooth muscle cells migrate into the lesion and secrete collagen, forming a fibrous cap over the lipid core; inflammatory cells (T cells, mast cells, macrophages) infiltrate the plaque; and calcium deposits develop in older plaques. The fibrous cap is the critical structural element — a thick, stable fibrous cap separates the thrombogenic lipid core from the flowing blood, maintaining plaque stability. A thin, eroded, or inflamed fibrous cap is vulnerable to rupture.
Plaque rupture exposes the lipid core to flowing blood, triggering platelet adhesion, activation, and aggregation on the thrombogenic surface and activating the coagulation cascade. The resulting thrombus can occlude the artery in situ (causing stroke from local thrombosis) or fragment and embolize distally (causing stroke from artery-to-artery embolism). This is identical to the mechanism of acute MI from coronary plaque rupture — atherosclerosis is a systemic disease affecting both the coronary arteries and the cerebral vasculature, which is why patients with known coronary artery disease have elevated stroke risk even without carotid stenosis, and vice versa.
Cholesterol Numbers — What Each Value Means for Stroke Risk
A standard lipid panel measures four primary values, each with distinct relevance to stroke risk:
LDL-C (low-density lipoprotein cholesterol) is the primary treatment target for atherosclerotic cardiovascular disease prevention. LDL carries approximately 70 percent of all plasma cholesterol and is the dominant driver of atherogenesis. The relationship between LDL-C and cardiovascular risk (including stroke) is log-linear and continuous — lower LDL is better at any starting level, with no established threshold below which further reduction stops providing benefit. Current secondary prevention guidelines recommend LDL-C below 70 mg/dL for most patients with ischemic stroke from atherosclerosis, with emerging evidence (from the FOURIER trial with evolocumab and the ODYSSEY OUTCOMES trial with alirocumab) supporting even lower targets (below 55 mg/dL) in very high-risk patients who tolerate aggressive PCSK9 inhibitor-based therapy.
HDL-C (high-density lipoprotein cholesterol) is a negative risk marker — higher HDL is associated with lower cardiovascular and stroke risk in observational studies. HDL particles are thought to facilitate reverse cholesterol transport (removing cholesterol from peripheral tissues and arterial walls back to the liver), and HDL has anti-inflammatory and antioxidant properties at the endothelial level. However, clinical trials testing drugs that raise HDL (niacin, CETP inhibitors) have not demonstrated cardiovascular event reduction — suggesting that HDL-C measured in plasma may be a biomarker of cardiovascular health rather than a causal protective factor. No drug treatment targeting HDL elevation has proven benefit for stroke prevention; the emphasis remains on LDL reduction.
Triglycerides contribute to stroke risk both directly (very high triglycerides, above 400 to 500 mg/dL, are associated with small, dense LDL particles that are particularly atherogenic) and indirectly (as a marker of insulin resistance, obesity, and metabolic syndrome — conditions that increase stroke risk through multiple pathways). Elevated triglycerides (150 to 499 mg/dL) are addressed primarily through lifestyle modification (reduced carbohydrate and alcohol intake, weight loss, physical activity) and diabetes management; triglyceride-specific medication (fibrates, omega-3 fatty acids) is reserved for very high triglyceride levels primarily to prevent pancreatitis, with uncertain benefit for cardiovascular event reduction.
Non-HDL-C (total cholesterol minus HDL-C) captures all atherogenic lipoproteins — LDL, IDL, VLDL, and Lp(a) — in a single composite measure. Non-HDL-C is a superior predictor of cardiovascular events compared to LDL-C in patients with elevated triglycerides (where LDL-C is often underestimated by standard Friedewald calculation) and is increasingly used as a co-primary treatment target alongside LDL-C in updated guidelines.
Statin Therapy for Stroke Prevention — The SPARCL Evidence
The landmark trial establishing statin therapy specifically for secondary stroke prevention is the SPARCL trial (Stroke Prevention by Aggressive Reduction in Cholesterol Levels), published in 2006. SPARCL enrolled 4,731 patients with ischemic stroke or TIA and no known coronary artery disease, randomized them to atorvastatin 80 mg versus placebo, and followed them for a median of 4.9 years. The results demonstrated a 16 percent relative reduction in fatal or nonfatal stroke (absolute risk reduction 2.2 percent), a 35 percent relative reduction in major cardiovascular events, and a significant 42 percent relative reduction in the risk of carotid revascularization in the atorvastatin group.
The SPARCL trial also identified an unexpected finding: a modestly increased risk of hemorrhagic stroke in the atorvastatin group (55 vs 33 events; HR 1.66, 95% CI 1.08 to 2.55). This finding — combined with the earlier epidemiological suggestion that low cholesterol might increase hemorrhagic stroke risk — has generated clinical debate, but is generally interpreted as not changing the favorable net clinical benefit of statin therapy after ischemic stroke given the much larger absolute reduction in ischemic stroke events. Current guidelines do not recommend withholding statins after ischemic stroke based on hemorrhagic stroke concern, but patients with prior hemorrhagic stroke (particularly those with cerebral amyloid angiopathy) warrant individualized risk-benefit assessment.
Beyond SPARCL, the CTT (Cholesterol Treatment Trialists) Collaboration meta-analysis of over 170,000 patients in 26 statin trials established that each 1 mmol/L (approximately 39 mg/dL) reduction in LDL-C reduces major vascular events (including stroke) by approximately 21 percent, with benefit proportional to the magnitude of LDL reduction. This dose-response relationship supports the use of high-intensity statins — atorvastatin 40 to 80 mg or rosuvastatin 20 to 40 mg — rather than moderate-intensity statins as the standard of care for secondary prevention after ischemic stroke.
Beyond Statins — PCSK9 Inhibitors and the Residual Risk Question
Despite high-intensity statin therapy, a substantial proportion of patients with ischemic stroke from atherosclerosis have residual cardiovascular risk — partly because statins typically reduce LDL by 50 percent (leaving LDL above target in patients with very high baseline LDL) and partly because other atherogenic lipoproteins (Lp(a), remnant particles) contribute to residual risk beyond LDL.
PCSK9 inhibitors (evolocumab and alirocumab) — injectable monoclonal antibodies that block PCSK9, the protein that degrades LDL receptors — reduce LDL by an additional 50 to 60 percent on top of maximally tolerated statin therapy, achieving LDL levels of 20 to 40 mg/dL in most patients. The FOURIER trial (evolocumab) and ODYSSEY OUTCOMES trial (alirocumab) both demonstrated significant cardiovascular event reduction when added to background statin therapy in very high-risk patients, including stroke reduction specifically. In the FOURIER trial, evolocumab reduced stroke by 25 percent compared to statin plus placebo. PCSK9 inhibitors are currently indicated for very high-risk patients (including prior ischemic stroke with established atherosclerotic cardiovascular disease) who are unable to reach LDL targets on maximum tolerated statin therapy.
Lipoprotein(a) — Lp(a) — is an emerging area of focus in stroke prevention. Lp(a) is genetically determined, largely independent of diet and lifestyle, and associated with increased stroke risk independently of LDL. Statins do not meaningfully lower Lp(a), and no approved therapy specifically targeting Lp(a) exists yet — though RNA-interference therapies (pelacarsen) are in late-phase clinical trials. Current guidelines recommend Lp(a) measurement at least once in adults with premature or familial atherosclerotic cardiovascular disease as a risk modifier in treatment decisions.
The American Stroke Association cholesterol and stroke resource explains the link between cholesterol management and stroke prevention. The CDC cholesterol risk factors page covers lipid panel interpretation and cardiovascular risk. The NHLBI cholesterol treatment guide reviews statin therapy, LDL targets, and lifestyle modifications for lipid management.
Related reading: What Is a Stroke? | High Blood Pressure and Stroke | Ischemic vs Hemorrhagic Stroke | Atrial Fibrillation | Coronary Artery Disease
Sources
- Amarenco P, et al. High-Dose Atorvastatin after Stroke or Transient Ischemic Attack (SPARCL). N Engl J Med. 2006;355(6):549-559.
- Cholesterol Treatment Trialists’ Collaboration. Efficacy and Safety of LDL-Lowering Therapy: Meta-analysis of Individual Data from 174,000 Participants. Lancet. 2010;376(9753):1670-1681.
- Sabatine MS, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease (FOURIER). N Engl J Med. 2017;376(18):1713-1722.
- Grundy SM, et al. 2018 AHA/ACC Guideline on the Management of Blood Cholesterol. J Am Coll Cardiol. 2019;73(24):e285-e350.
- Libby P, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5(1):56.
Lifestyle Modification for Cholesterol and Stroke Risk Reduction
Lipid-lowering medications — primarily statins and PCSK9 inhibitors — are the most potent tools for LDL reduction and stroke prevention, but lifestyle modifications provide meaningful additional benefit, address non-LDL risk factors (triglycerides, inflammation, insulin resistance), and are essential complements to pharmacotherapy:
Dietary fat modification: Replacing saturated fats (found in red meat, full-fat dairy, and tropical oils) with unsaturated fats (olive oil, avocado, nuts, fatty fish) reduces LDL cholesterol by approximately 5 to 15 percent and favorably modifies the LDL particle composition (reducing small, dense LDL particles that are particularly atherogenic). Trans fats — produced by partial hydrogenation of vegetable oils and found in processed and fried foods — raise LDL and lower HDL simultaneously, and their elimination from the food supply (now mandatory in the US and many other countries) has been associated with measurable reductions in cardiovascular event rates. The PREDIMED trial established that the Mediterranean diet — emphasizing olive oil, nuts, vegetables, fruits, legumes, and fish — reduces stroke risk by 31 to 33 percent compared to a low-fat control diet, an effect that extends beyond LDL reduction to include anti-inflammatory, anti-thrombotic, and endothelial protective mechanisms.
Dietary cholesterol: Dietary cholesterol (from eggs, shellfish, organ meats) has a more modest and variable effect on LDL-C than dietary saturated fat — most people are “cholesterol compensators” whose intestinal cholesterol absorption and hepatic cholesterol synthesis adjust to maintain relatively stable serum cholesterol regardless of dietary cholesterol intake. Approximately 25 percent of people are “cholesterol hyperabsorbers” in whom dietary cholesterol has a greater effect on serum LDL. Current dietary guidelines have relaxed specific numerical limits on dietary cholesterol in favor of emphasizing overall dietary pattern quality (Mediterranean, DASH), where total saturated fat intake, dietary fiber, and plant sterol content have larger effects on LDL than cholesterol intake alone.
Dietary fiber: Soluble fiber (from oats, barley, beans, lentils, psyllium, and many fruits and vegetables) reduces LDL-C by 5 to 10 percent through two mechanisms: binding bile acids in the intestinal lumen, reducing their reabsorption and requiring the liver to convert more cholesterol to bile acids; and providing fermentation substrate for gut microbiota that produce short-chain fatty acids inhibiting hepatic cholesterol synthesis. The USFDA has permitted a health claim for beta-glucan (the specific soluble fiber in oats and barley) reducing cardiovascular disease risk based on clinical trial evidence. Increasing dietary fiber intake is one of the most accessible and broadly beneficial dietary changes for LDL reduction with no adverse effects.
Physical activity: Regular aerobic exercise reduces triglycerides by 15 to 20 percent, modestly increases HDL-C by 3 to 6 percent, shifts LDL particle distribution toward larger, less atherogenic particles, and reduces LDL oxidation. The effect of exercise on LDL-C itself is relatively modest (approximately 5 percent reduction), but the cumulative impact on cardiovascular risk through lipid-independent pathways (blood pressure reduction, insulin sensitivity improvement, reduced inflammation, improved endothelial function) makes physical activity a cornerstone of cardiovascular risk reduction beyond its direct lipid effects.
Weight management: Obesity — particularly central adiposity — is associated with elevated triglycerides, low HDL, and increased small dense LDL. A 5 to 10 percent weight reduction in overweight individuals typically reduces triglycerides by 20 percent and increases HDL by 5 to 10 percent, while also improving insulin resistance and blood pressure — multiplying the stroke risk reduction benefit beyond the lipid changes themselves. Weight reduction through a caloric deficit combined with increased physical activity is more effective for dyslipidemia management than either intervention alone.
Statin Intolerance — When the Standard Treatment Is Not Tolerated
Statin-associated muscle symptoms (SAMS) — ranging from mild myalgia (muscle pain or weakness without CK elevation) to the rare but serious myopathy and rhabdomyolysis — are the most common reason patients stop statin therapy. Reported rates of SAMS in clinical practice (5 to 20 percent) substantially exceed the rates seen in randomized controlled trials (approximately 1 to 5 percent), suggesting that a significant portion of symptoms attributed to statins are not pharmacologically caused — the placebo-controlled SAMSON trial demonstrated that 90 percent of statin-attributed symptoms also occurred with placebo, and the nocebo effect (patient expectation of side effects) is a substantial contributor to statin discontinuation.
For patients with true statin intolerance (confirmed by objective CK elevation or by symptoms that resolve with drug discontinuation and recur with rechallenge), several strategies maintain stroke prevention benefit:
Switching to a different statin at lower dose — myopathy risk varies substantially across statins, and a patient who cannot tolerate simvastatin at high dose may tolerate rosuvastatin or pravastatin well. Alternate-day dosing of long half-life statins (rosuvastatin, atorvastatin) can maintain meaningful LDL reduction with fewer statin-days and reduced symptom burden. Ezetimibe (which inhibits intestinal cholesterol absorption rather than hepatic synthesis) reduces LDL by an additional 15 to 20 percent as a standalone agent and by 20 to 25 percent when added to maximally tolerated low-dose statin — with an excellent tolerability profile and no muscle-related effects. The IMPROVE-IT trial established that ezetimibe plus simvastatin significantly reduced major cardiovascular events including stroke compared to simvastatin alone, providing the first non-statin evidence for LDL-lowering-mediated cardiovascular benefit. For patients unable to tolerate any statin at any dose, PCSK9 inhibitors provide the most potent LDL reduction alternative with a safety profile unrelated to statin myopathy.
Cholesterol Management After Ischemic Stroke — A Practical Framework
For patients who have experienced an ischemic stroke or TIA attributable to atherosclerosis, the following clinical framework reflects current evidence-based practice:
All patients should receive high-intensity statin therapy (atorvastatin 40 to 80 mg or rosuvastatin 20 to 40 mg) regardless of baseline LDL — the SPARCL trial demonstrated benefit even in patients with baseline LDL below 100 mg/dL, and guidelines do not identify an LDL level below which statin therapy is not beneficial for secondary prevention after ischemic stroke from atherosclerosis. A fasting lipid panel should be checked 4 to 6 weeks after initiating or escalating therapy to assess response and allow dose adjustment. LDL targets are below 70 mg/dL for most patients and below 55 mg/dL for very high-risk patients (multiple atherosclerotic events or atherosclerotic events with multiple high-risk conditions).
If the LDL target is not achieved with maximum tolerated statin therapy, add-on therapy should be initiated: ezetimibe is the preferred first add-on based on the IMPROVE-IT trial data, tolerability, and cost. If the target remains unmet on statin plus ezetimibe, PCSK9 inhibitor therapy (evolocumab or alirocumab) is indicated and supported by guidelines for very high-risk secondary prevention patients. The combination of high-intensity statin plus ezetimibe plus PCSK9 inhibitor can reduce LDL to below 20 to 30 mg/dL in most patients — levels where further atherosclerotic plaque regression has been demonstrated by imaging studies.
Beyond LDL, a complete atherosclerotic risk factor profile should be optimized simultaneously: blood pressure to below 130/80 mmHg, smoking cessation, diabetes management to HbA1c below 7 percent, antiplatelet therapy (aspirin plus clopidogrel for the first 21 to 90 days after stroke, then single antiplatelet long-term for non-cardioembolic stroke), and carotid imaging to identify high-grade stenosis that may warrant carotid endarterectomy. Addressing all modifiable risk factors simultaneously rather than sequentially provides the greatest absolute stroke risk reduction — because the combination of controlled blood pressure, optimized LDL, and antiplatelet therapy reduces recurrent stroke risk by 80 to 85 percent compared to no secondary prevention, reflecting the multiplicative benefit of multiple concurrent interventions.